


Several subscribers have asked me to comment on the new classification system for multiple sclerosis (MS) published last month in the journal Brain, which uses MRI and serum neurofilament levels. One person asked whether this means MS is now two diseases rather than one. Other commentators have hinted that this will change how we diagnose, treat and manage MS.

In short, none of these claims are supported by the research findings presented in this paper (Willard et al. Combined magnetic resonance imaging and serum analysis reveals distinct multiple sclerosis types. Brain. 2025 Dec 4;148(12):4578-4591). The article introduces a new machine learning model that integrates MRI data with serum neurofilament light chain (sNfL) levels to supposedly categorise MS based on ‘biology’ rather than ‘MS-related symptoms’. The assumption that MS-related symptoms are not biological is incorrect. The fact that humans, and by inference, people with MS, are biological machines means their symptoms are biological.
The Oxford dictionary defines biology as ‘the study of living organisms, divided into many specialised fields that cover their morphology, physiology, anatomy, behaviour, origin, and distribution.’ Symptoms of a disease are hence biological.
The study combined fluid biomarkers (sNFL) with brain imaging, which then identified two distinct disease subtypes: early-sNfL and late-sNfL. The early-sNfL group is characterised by higher levels of inflammatory activity, significant lesion accrual, and faster brain atrophy, whereas the late-sNfL group exhibits more gradual neurodegeneration. The authors claim the multimodal approach, including sNFL, proved superior to MRI-only models in correlating with patient disability and predicting how individuals respond to therapeutic interventions. They claim the findings may provide a prognostic framework to support personalised medicine by identifying more aggressive disease states earlier in the clinical course, which are more likely to predict treatment response.
Limitations
Please note that the authors have identified several limitations and flaws in their study, which can be categorised into issues with the study population, methodology, and challenges related to clinical implementation. The study utilised data drawn from clinical trial cohorts, which means the participants do not fully represent the broader MS population. Due to strict eligibility criteria in the source trials, the study lacks data on underrepresented ethnic groups and patients with co-morbidities. While the model was trained on a cohort including both relapsing–remitting and secondary progressive MS, the external validation was conducted only on a cohort of newly diagnosed patients (early MS). Consequently, the model’s accuracy for late-stage disease remains to be validated.
The external test dataset had a limited range of EDSS scores, which likely contributed to lower correlation coefficients between the model stages and disability measures in the test set than in the training set. Although the study aimed to use unsupervised machine learning, the pipeline was “not entirely unsupervised” because the initial feature selection step relied on correlations with the EDSS. The researchers noted that the decision to narrow the selection to exactly five variables was an “arbitrary decision” guided by the dataset’s size and resources. The SuStaIn algorithm assumes that disease progression follows a “monotonic sequence,” in which subtypes accumulate abnormalities in a fixed order. This assumption facilitates modelling but may limit the model’s sensitivity to fluctuating disease trajectories. In the longitudinal analysis, sNfL levels decreased in untreated control subjects. The authors attribute this to “regression to the mean,” as patients were recruited during active phases of inflammation (a requirement for trial entry), which naturally subsided, complicating the assessment of actual therapeutic effects.
The authors acknowledge that few hospitals currently possess the necessary infrastructure to convert routine MRI scans into the precise quantitative measures required by their model. Quantitative MRI measures are sensitive to differences in scanners and acquisition parameters. While the study used harmonisation (the ComBat algorithm) to mitigate this, such harmonisation poses practical challenges for widespread clinical adoption. The correlations between the model-derived stages and the EDSS were weak. The authors argue this is expected because EDSS is weighted toward motor function, while MRI/sNfL changes often precede clinical symptoms. Still, it highlights a gap between the biological staging and current clinical disability measures. To ensure the model remained accessible for potential clinical translation, the study excluded more advanced imaging modalities (such as myelin-sensitive MRI sequences), which might have provided more comprehensive insights.
I am clearly not on the same page as the authors.
My initial thoughts
The title refers to distinct MS subtypes. Use of the term “distinct" is a misnomer, as some subjects switched from one subtype to the other.
“Given that 7% of patients switched from one subtype to another in the training dataset, and 23% switched in the testing dataset, these subtypes are likely to represent a continuum of underlying pathology.”
The title is therefore misleading and argues against two distinct subtypes of MS.
Please note that the training dataset was derived from a phase 2 Evobrutinib clinical trial conducted between March 2017 and July 2018. The study subjects were selected using well-defined inclusion and exclusion criteria. All trial subjects had to have one or more documented relapses within the 2 years before screening, with either one relapse which occurred within the last year before randomisation or the presence of at least one T1 gadolinium-enhancing lesion within 6 months before randomisation—about a quarter of study subjects had been exposed to DMTs in the past.
Therefore, the cohort used for training and model development had established active MS, as defined by relapses and/or Gd-enhancing lesions on MRI. Another issue is that they were diagnosed using the 2011 McDonald criteria. This creates problems, as subjects with active MS tend to become less active over time due to regression to the mean. In addition, the consequences of having active MS will then unfold over time as part of the natural history of MS. I would be interested to know how the model would have been developed if it included pwMS who did not have active MS as defined by the trial inclusion criteria. I suspect very differently.
Is the model simply measuring baseline MS disease activity, including raised sNFL as an activity marker, and then predicting the consequences of this period of MS disease activity on the end organ, i.e., brain volume loss or end-organ damage? In comparison, those without activity at baseline, i.e. a normal sNFL, will not have the same trajectory in terms of the pathology in the end-organ as measured by MRI, but are likely to regress to the mean in the opposite direction; i.e. have a delayed rise in sNFL as part of the fluctuation in MS disease activity over time. I think any model of MS, a dynamic disease, needs more than a few limited MRI metrics and sNFL to capture its behaviour.
In comparison to the training dataset, the testing dataset was from the phase 3 REFLEX trial that compared two dosing frequencies of subcutaneous interferon beta-1a in patients with a first clinical demyelinating event suggestive of multiple sclerosis or CIS. These subjects were diagnosed using the older 2005 McDonald criteria, were younger and were naive to DMTs. The subjects in this study had CIS rather than established MS. I know that a subsequent analysis of the REFLEX clinical trial population, retrospectively applying the McDonald 2017 MS diagnostic criteria, estimated that about 50% of these subjects would have been classified as having MS. This means that 50% didn’t fulfill the requirements for MS and are more likely to have benign MS. Validating the model developed on established active MS using an early, much younger group of subjects naive to DMTs diagnosed with different diagnostic criteria makes little sense to me, i.e. it is flawed from a scientific perspective, and is likely to introduce bias. For example, the validation using subjects very early in the course of their disease, who were much younger and hence had a greater ability to recover function and repair damage, introduces a biological variable not measured by the model. How does neurological reserve and recovery of function impact the model?
An essential aspect of clinical outcomes and various biomarkers in MS is that they evolve at different rates. In other words, the temporal sequence of how they change over time is out of sequence with each other. I have referred to this phenomenon in the past as lag. Let me give you some examples. Demyelination develops along a specific pathway before loss of function and before a Gd-enhancing lesion is seen. Changes in the magnetisation transfer ratio (MTR) on MRI can occur in normal-appearing white matter (NAWM) several months before an MS lesion becomes visible with Gd-enhancement. This indicates that MTR changes are earlier and more sensitive markers of pre-lesional tissue damage than Gd enhancement. At some point, axonal injury occurs, leading to NFL release. Based on serial sampling studies, we suspect this process may precede the onset of clinical relapse symptoms and the detection of Gd enhancement on MRI. However, these latter processes are likely to cluster into a relatively narrow window that lasts days to weeks. We know that Gd enhancement of a lesion typically lasts 2-3 weeks before resolving, whereas NFL levels remain elevated for months. The latter occurs because of Wallerian degeneration, which takes a long time to unfold: it can take months to clear the debris from transected axons. The proximal axonal degeneration above the lesion may take even longer and play out over years. Similarly, repaired or remyelinated axons may be programmed to die off in the future. We think they are vulnerable to early ageing, energy failure and delayed excitotoxicity. This delayed neurodegeneration may happen over decades. I try to illustrate these time changes in this cartoon that I made more than a decade ago.
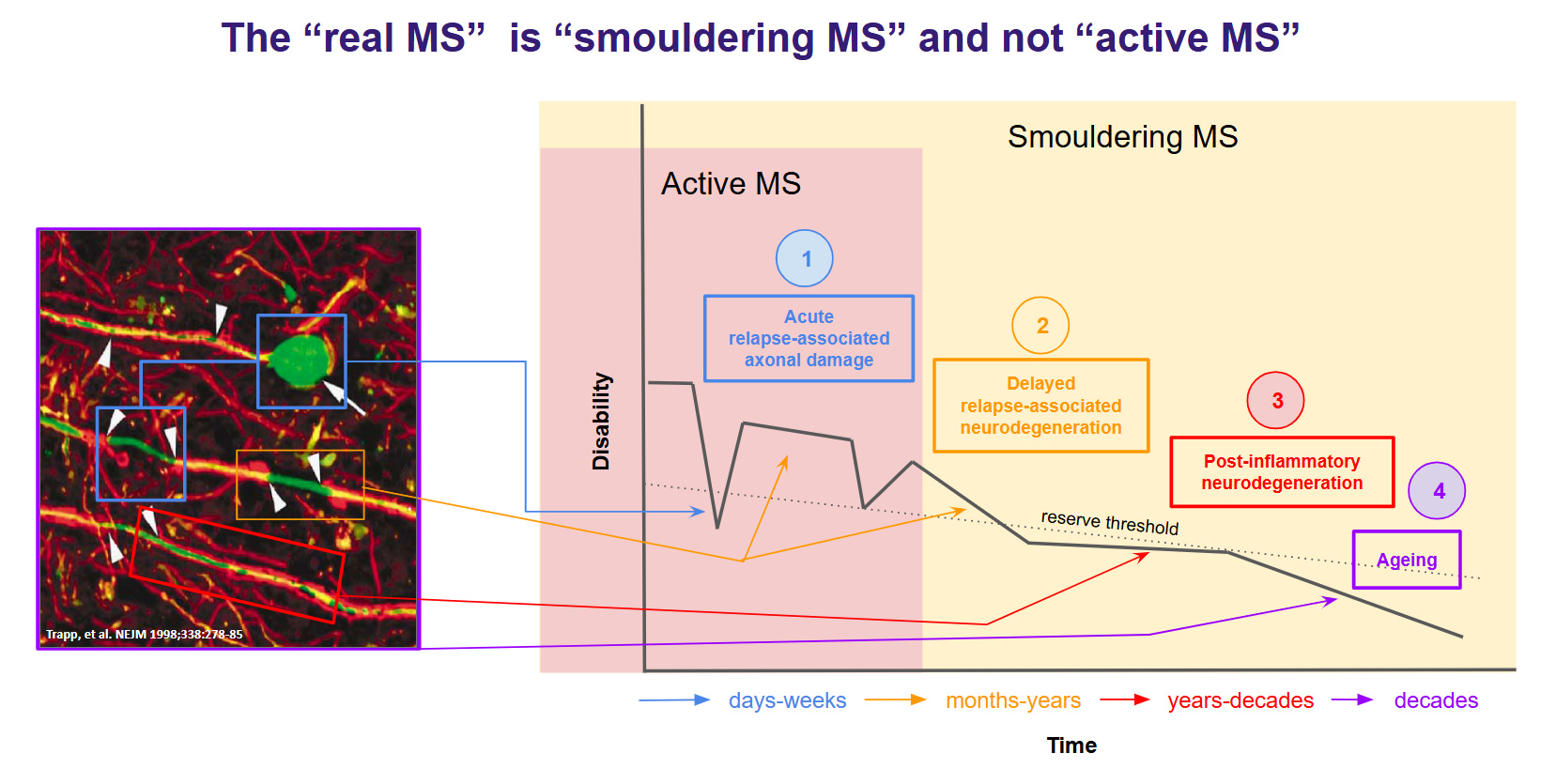
The loss of tissue from an acute lesion, i.e., the subsequent atrophy, can take months to years to occur. In optic neuritis and the optic nerve model, atrophy is seen after 3 months and reaches a plateau at about 6 months. Therefore, the inflammation detected now, as evidenced by Gd-enhancing lesions and/or elevated NFL levels, will result in whole-brain or regional atrophy six or more months later. With longer axons than those in the optic nerve, it will take longer than 6 months to reach a plateau. The point I am making is that I don’t know how this model accounts for the lag in changes in these biomarkers. The study utilises five specific MRI-derived measures, three of which are volume measures (limbic cortex, deep grey matter, and parietal cortex volume), all of which are likely to be impacted by lag.
The other two MRI metrics are the total T2 lesion volume and the corpus callosum white matter T1-weighted/T2-weighted ratio. I am aware that the T2 lesion volume changes with time at an individual lesion level in response to treatment. In general, the T2 lesion volume of an acute MS lesion is large and decreases as the Gd-enhancement disappears, i.e., the lesion shrinks in size. Most MRI analyses show a reduction in T2 volumes with treatment. Therefore, this component of the model would be affected by treatment.
The T1-weighted/T2-weighted (T1w/T2w) ratio in the white matter of the corpus callosum changes significantly over time as a function of age. After middle age, the T1w/T2w ratio in the white matter and corpus callosum generally begins to decline. This decrease is thought to be associated with age-related microstructural changes, including myelin degeneration and loss of white matter integrity. How do the significant age differences between the training and validation datasets affect this component of the model?
Therefore, these MRI metrics are not static and change over time. How these dynamic changes affect the model is unknown. The changes may not be that important at a group level. However, I suspect they will create a lot of noise or variability in individual datasets, which may make it difficult to use for decision-making at a patient level.
Summary
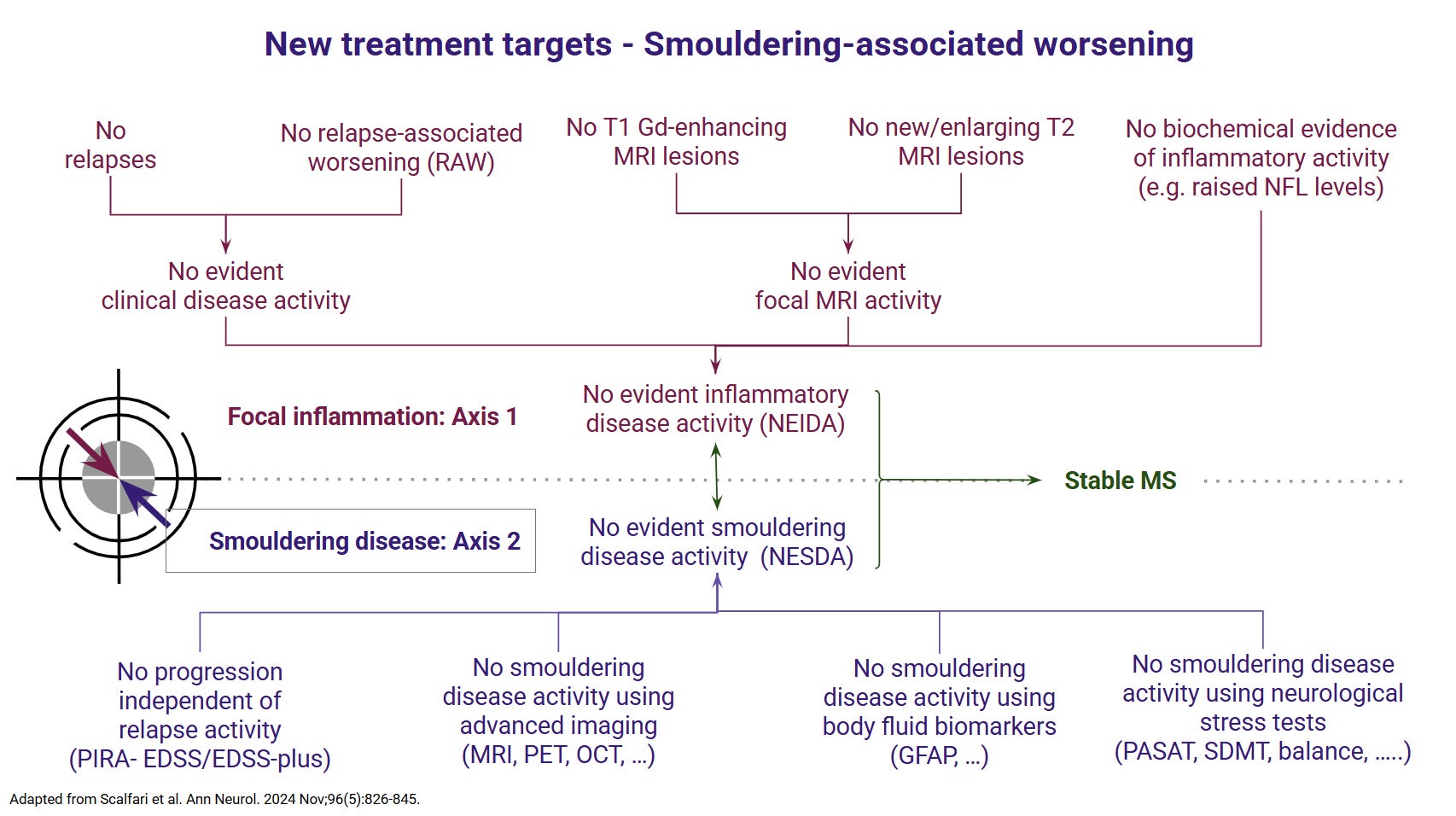
A crucial point made by the authors in the discussion is that few centres have the infrastructure to reliably analyse the scans and generate the metrics required as inputs into this model. Therefore, it isn't easy to see how this model will impact precision medicine. In comparison, sNFL and CSF NFL levels are beginning to enter routine clinical practice and are increasingly being used to aid in clinical decision-making. To be blunt, I am not sure how this paper changes my thinking about MS and its management. Our therapeutic strategies remain the same: early, effective treatment is the best way to protect the end organ. Our treatment targets remain no evident inflammatory disease activity (NEIDA) and no evident smouldering disease activity (NESDA), regardless of what proposed subtype of MS you have. We have very effective treatments for NEIDA and less effective treatments for NESDA; it would take a brave neurologist not to treat a person with a low-sNFL phenotype with an anti-inflammatory. Similarly, it would not make sense to ignore smouldering MS in a person with a high sNFL phenotype. The challenge from now on for the MS community is developing treatments for smouldering MS to achieve stable MS and long-term remission regardless of putative subtypes.
Does my response to this paper make sense? I am prepared to answer further questions.
Paper
Multiple sclerosis (MS) is a highly heterogeneous disease in its clinical manifestation and progression. Predicting individual disease courses is key for aligning treatments with underlying pathobiology. We developed an unsupervised machine learning model integrating MRI-derived measures with serum neurofilament light chain (sNfL) levels to identify biologically informed MS subtypes and stages. Using a training cohort of patients with relapsing-remitting and secondary progressive MS (n = 189), with validation on a newly diagnosed population (n = 445), we discovered two distinct subtypes defined by the timing of sNfL elevation and MRI abnormalities (early- and late-sNfL types). In comparison to MRI-only models, incorporating sNfL with MRI improved correlations of data-derived stages with the Expanded Disability Status Scale in the training (Spearman’s ρ = 0.420 versus MRI-only ρ = 0.231, P = 0.001) and external test sets (ρ = 0.163 for MRI-sNfL, versus ρ = 0.067 for MRI-only). The early-sNfL subtype showed elevated sNfL, corpus callosum injury and early lesion accrual, reflecting more active inflammation and neurodegeneration, whereas the late-sNfL group showed early volume loss in the cortical and deep grey matter volumes, with later sNfL elevation. Cross-sectional subtyping predicted longitudinal radiological activity: the early-sNfL group showed a 144% increased risk of new lesion formation (hazard ratio = 2.44, 95% confidence interval 1.38-4.30, P < 0.005) compared with the late-sNfL group. Baseline subtyping, over time, predicted treatment effect on new lesion formation on the external test set (faster lesion accrual in early-sNfL compared with late-sNfL, P = 0.01), in addition to treatment effects on brain atrophy (early sNfL average percentage brain volume change: -0.41, late-sNfL = -0.31, P = 0.04). Integration of sNfL provides an improved framework in comparison to MRI-only subtyping of MS to stage disease progression and inform prognosis. Our model predicted treatment responsiveness in early, more active disease states. This approach offers a powerful alternative to conventional clinical phenotypes and supports future efforts to refine prognostication and guide personalized therapy in MS.
Accidental readers
If you have been forwarded this email and are not an MS-Selfie subscriber, please consider subscribing and helping MS-Selfie expand its resources for the broader MS community. MS-Selfie relies on subscriptions to fund its curated MS-Selfie microsite, MS-Selfie books, MS-Selfie Infocards, and other activities that extend beyond the MS-Selfie Substack newsletters.
Subscriptions and donations
MS-Selfie newsletters and access to the MS-Selfie microsite are free. In comparison, off-topic Q&A sessions are restricted to paying subscribers. Subscriptions are being used to run and maintain the MS Selfie microsite and other related activities, as I don’t have time to do this myself. You must be a paying subscriber to ask questions unrelated to the newsletters or podcasts. If you can’t afford to become a paying subscriber, please email a request for a complimentary subscription (ms-selfie@giovannoni.net).
Questions
If you have questions unrelated to the newsletters or podcasts, please email them to ms-selfie@giovannoni.net. Prof. G will try to answer them as quickly as possible.
Important Links
🖋 Medium
General Disclaimer
Please note that the opinions expressed here are those of Professor Giovannoni and do not necessarily reflect the positions of Queen Mary University of London or Barts Health NHS Trust. The advice is intended as general and should not be interpreted as personal clinical advice. If you have problems, please tell your healthcare professional, who will be able to help you.






