



There are too many ECTRIMS 2024 highlights to cover in one newsletter. However, the top of the list of highlights was the positive tolebrutinib results in non-relapsing (inactive) secondary progressive MS. At the bottom was the negative, but not unexpected, simvastatin trial in SPMS.

Tolebrutinib
Tolebrutinib, a second-generation BTK inhibitor (BTKi), has a dual mode of action in MS. It inhibits BTK in B-cells and is an anti-inflammatory therapy. In both the relapsing and non-relapsing progressive MS trials, it had a relatively modest impact on relapses and focal MRI activity and more or less matched the anti-inflammatory effects of evobrutinib, the first BTKi (see paper below). Both tolebrutinib and evobrutinib were not shown to be superior to teriflunomide in suppressing relapse and focal Gd-enhanced MRI activity.

Teriflunomide, however, was superior to tolebrutinib in suppressing Gd-enhancing lesions but not new T2 lesions. This would indicate that new Gd-enhancing lesions are less likely to form chronic T2 lesions in subjects treated with tolebrutinib. This suggests that tolebrutinib is changing the biology of how MS lesions evolve; i.e. it is doing something to the acute Gd-enhancing lesions that makes it less likely to leave a scar or new T2 lesion. Based on this preliminary observation, I suspect that in addition to fewer T2 lesions, tolebrutinib will also suppress the development of paramagnetic rim (PRLs) and slowly expanding lesions (SELs), both associated with worse outcomes. These data have yet to be analysed. It will be interesting to see if these MRI findings also apply to blood neurofilament levels, i.e. if new Gd-enhancing lesions are less likely to form new T2 lesions on tolebrutinib subjects on tolebrutinib should have lower NFL levels associated with Gd-enhancing scans than those occurring in patients on teriflunomide or placebo.

However, despite being inferior to teriflunomide in suppressing focal inflammatory activity, tolebrutinib was superior to it when it came to disability progression., i.e. it has a robust impact on smouldering MS-associated worsening (SAW) but not relapses.
Tolebrutinib, therefore, dissociates relapses from disability progression and proves that these two processes are likely to be independent of each other with the caveat that relapses can be associated with some disability progression called relapse-associated worsening or RAW. This treatment effect on smouldering MS was seen in the relapsing (Gemini 1&2 trials) and non-relapsing SPMS trials (Hercules trial). Subjects in the Hercules trial were also more likely to have significant disability improvement, with a trend in the relapsing or Gemini trials. The disability improvement data is critical and tells us that something is happening centrally to promote recovery of function in subjects on tolebrutinib. I hope biomarker data can support the recovery of function data.

How do you explain tolebrutinib’s effect on smouldering MS and its moderate impact on focal inflammatory activity? Tolebrutinib is a CNS penetrant BTKi and will inhibit CNS resident B-cells and plasmablasts. In addition, it downregulates and inhibits microglia and macrophages via the so-called Fc-gamma III receptor. So, this agent tackles some CNS processes that drive smouldering MS and may switch microglia into a regulatory phenotype that promotes remyelination and recovery of function.

Notably, the data on reduced disability progression and recovery of function on tolebrutinib was not associated with a significant impact on brain volume loss. This needs to be explained. Brain volume is a complex measure, and a shrinking in the volume of microglia may confound any protection of volume by reduced neuroaxonal loss. Therefore, we will have to wait for more detailed analyses of regional brain volume and other MRI metrics from these studies to interpret this data. What is clear is that simply using brain volume alone as a biomarker does not explain what is happening at the tissue level.
Please note that tolebrutinib and the other BTKIs are associated with liver toxicity. Hence, if they get to the clinic, they will require frequent blood monitoring for the first 3-6 months to prevent severe drug-induced liver injury (DILI).
What about other BTK’s?
The phase 2 extension data of fenebrutinib, another BTKi in phase 3, was presented as an ePoster. Compared to tolebrutinib, fenebrutinib is much more effective at suppressing Gd-enhancing lesions. Based on this data, I would give fenebrutinib a ~87.5% (range 75-95%) of being superior to teriflunomide in suppressing relapses and focal MRI activity. The billion-dollar question is whether or not it will have an impact on smouldering MS pathology to a similar degree to tolebrutinib.
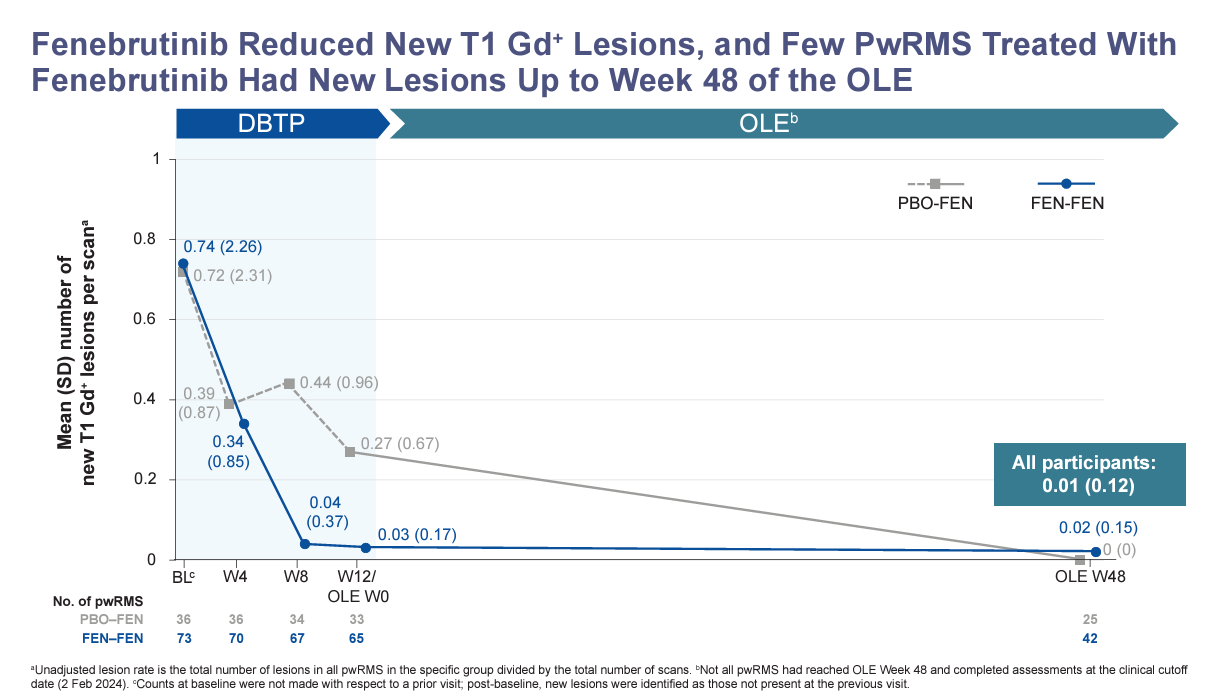
I raised this question with a pharmacologist and got the following response.
‘Fenebrutinib differs from tolebrutinib in that fenebrutinib is a non-covalent reversible inhibitor that targets the “open” conformation of the enzyme and acts to trap BTK in an inactive conformation. Fenebrutinib is reasonably potent, more so than evobrutinib, but about 9-fold less potent than tolebrutinib. From a PK perspective, tolebrutinib has a very short half-life (~90 min), while fenebrutinib is ~4-6 hrs. Because tolebrutinib is covalent, the PD actions are long-lasting, and enzyme activity recovers after the last dose within 5-7 days as new enzyme is synthesized (protein turnover). Fenebrutinib is a classical competitive inhibitor, so to maintain “coverage” in vivo, relatively high doses are needed, given twice daily. Fenebrutinib has a “long residency time”, meaning that the off-rate is slow. The consequence of nanomolar potency with slow dissociation means that the steady-state is defined by the off-rate. When measured in a model system, occupancy of BTK within cells is driven primarily by C trough, with estimates being that fenebrutinib is likely to have modest occupancy within the CNS. Interestingly, remibrutinib is also a hybrid covalent inhibitor that targets the same open conformation as fenebrutinib but also reacts with the C481 residue to form a thioether adduct like tolebrutinib evobrutinib, ibrutinib, etc.’
The proof will be in the trial results. You may be interested in knowing that fenebrutinib is being compared to ocrelizumab in a primary progressive trial. Ocrelizumab is setting a high bar, and if fenebrutinib is superior to ocrelizumab in PPMS, this will further prove the need to target CNS mechanisms to slow down SAW.
After deep thinking about this data this weekend, I will be optimistic and give fenebrutinib a 40% chance (range 30-50%) of being superior to ocrelizumab in PPMS. The latter is based on its superior anti-inflammatory effect to tolebrutinib and the fact that it does get into the CNS at a level likely to inhibit BTK in both B-cells and microglia. If I am proven correct about fenebrutinib, this would be great news for pwPPMS.
The dark horse is remibrutinib. We have no detailed PK and PD data on its CNS effects. However, the news on the ECTRIMS grapevine is that remibrutinib does not have a clear liver toxicity signal. If that is the case and it is as effective as tolebrutinib and fenebrutinib, it may just win the battle of the BTKi’s on safety.
How will the BTKi’s be used in clinical practice?
It is too early to speculate. But if the regulators’ license tolebrutinib to reduce SAW in non-active SPMS, it will be used as a monotherapy in more advanced MS. However, as SAW or PIRA occurs very early in the course of MS I suspect we will be identifying SAW early and switching patients early from other DMTs onto tolebrutinib in early MS. In other words, the diagnosis of SPMS will move to an earlier time in the course of MS. Going forward, tolebrutinib may be best used as part of an induction-maintenance protocol after an anti-CD20 or immune reconstitution therapy.
You can download my ECTRIMS slides from here.
Papers of interest
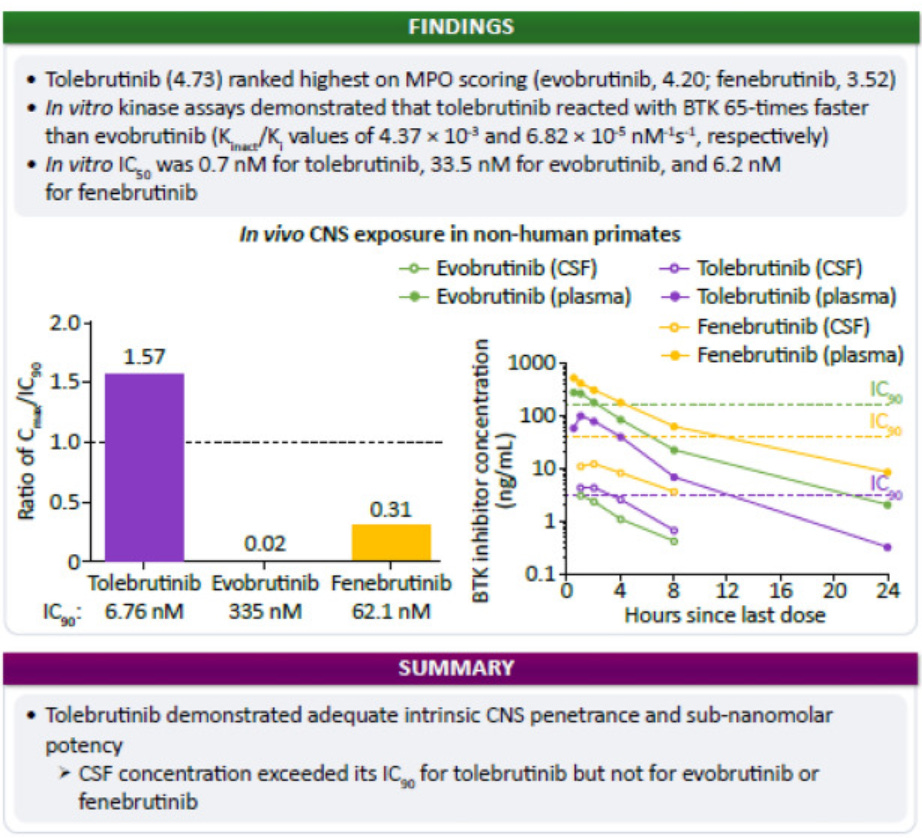
Background and objectives: Tolebrutinib is a covalent BTK inhibitor designed and selected for potency and CNS exposure to optimize impact on BTK-dependent signaling in CNS-resident cells. We applied a translational approach to evaluate three BTK inhibitors in Phase 3 clinical development in MS with respect to their relative potency to block BTK-dependent signaling and exposure in the CNS METHODS: We used in vitro kinase and cellular activation assays, alongside pharmacokinetic sampling of cerebrospinal fluid (CSF) in the non-human primate cynomolgus to estimate the ability of these candidates (evobrutinib, fenebrutinib, and tolebrutinib) to block BTK-dependent signaling inside the CNS.
Results: In vitro kinase assays demonstrated that tolebrutinib reacted with BTK 65-times faster than evobrutinib, while fenebrutinib, a classical reversible antagonist with a Ki value of 4.7 nM and slow off-rate (1.54 x 10-5 s-1), also had an association rate 1760-fold slower (0.00245 μM-1 * s-1). Estimates of cellular potency were largely consistent with the in vitro kinase assays, with an estimated IC50 of 0.7 nM for tolebrutinib against 33.5 nM for evobrutinib and 2.9 nM for fenebrutinib. We then observed that evobrutinib, fenebrutinib, and tolebrutinib achieved similar levels of exposure in non-human primate CSF after oral doses of 10 mg/kg. However, tolebrutinib CSF exposure (4.8 ng/mL) (kp,uu CSF=0.40) exceeded the IC90 (the estimated concentration inhibiting 90% of kinase activity) value, while evobrutinib (3.2 ng/mL) (kp,uu CSF=0.13) and fenebrutinib (12.9 ng/mL) (kp,uu CSF=0.15) failed to reach the estimated IC90 values.
Conclusions: Tolebrutinib was the only candidate of the three that attained relevant CSF exposure in non-human primates.
Background: Bruton's tyrosine kinase (BTK) regulates the functions of B cells and myeloid cells that are implicated in the pathogenesis of multiple sclerosis. Evobrutinib is a selective oral BTK inhibitor that has been shown to inhibit B-cell activation both in vitro and in vivo.
Methods: In this double-blind, randomized, phase 2 trial, we assigned patients with relapsing multiple sclerosis to one of five groups: placebo, evobrutinib (at a dose of 25 mg once daily, 75 mg once daily, or 75 mg twice daily), or open-label dimethyl fumarate (DMF) as a reference. The primary end point was the total (cumulative) number of gadolinium-enhancing lesions identified on T1-weighted magnetic resonance imaging at weeks 12, 16, 20, and 24. Key secondary end points included the annualized relapse rate and change from baseline in the score on the Expanded Disability Status Scale (EDSS).
Results: A total of 267 patients were randomly assigned to a trial group. The mean (±SD) total number of gadolinium-enhancing lesions during weeks 12 through 24 was 3.85±5.44 in the placebo group, 4.06±8.02 in the evobrutinib 25-mg group, 1.69±4.69 in the evobrutinib 75-mg once-daily group, 1.15±3.70 in the evobrutinib 75-mg twice-daily group, and 4.78±22.05 in the DMF group. The baseline adjusted rate ratios for the total number of lesions over time as compared with placebo were 1.45 in the evobrutinib 25-mg group (P = 0.32), 0.30 in the evobrutinib 75-mg once-daily group (P = 0.005), and 0.44 in the evobrutinib 75-mg twice-daily group (P = 0.06). The unadjusted annualized relapse rate at week 24 was 0.37 in the placebo group, 0.57 in the evobrutinib 25-mg group, 0.13 in the evobrutinib 75-mg once-daily group, 0.08 in the evobrutinib 75-mg twice-daily group, and 0.20 in the DMF group. There was no significant effect of trial group on the change from baseline in the EDSS score. Elevations in liver aminotransferase values were observed with evobrutinib.
Conclusions: Patients with relapsing multiple sclerosis who received 75 mg of evobrutinib once daily had significantly fewer enhancing lesions during weeks 12 through 24 than those who received placebo. There was no significant difference with placebo for either the 25-mg once-daily or 75-mg twice-daily dose of evobrutinib, nor in the annualized relapse rate or disability progression at any dose. Longer and larger trials are required to determine the effect and risks of evobrutinib in patients with multiple sclerosis. (Funded by EMD Serono; ClinicalTrials.gov number, NCT02975349.).
Additional reading
These studies’ results support my claim that relapses are not MS and that the real MS is smouldering MS. I have been saying this for some time. I suggest you read the following MS-Selfie Newsletters and papers to learn about some of the issues these trial results raise and support.
If you use the search function on MS-Selfie and search for the term "smouldering", you will find many more newsletters, podcasts, and case studies on this topic, which becomes very pertinent now that we have emerging treatments that work on smouldering MS. I sincerely hope we have given the PPMS Tolebrutinib study enough power to deliver a positive result as well.
Using a philosophical approach or deductive reasoning, we challenge the dominant clinico-radiological worldview that defines multiple sclerosis (MS) as a focal inflammatory disease of the central nervous system (CNS). We provide a range of evidence to argue that the 'real MS' is in fact driven primarily by a smouldering pathological disease process. In natural history studies and clinical trials, relapses and focal activity revealed by magnetic resonance imaging (MRI) in MS patients on placebo or on disease-modifying therapies (DMTs) were found to be poor predictors of long-term disease evolution and were dissociated from disability outcomes. In addition, the progressive accumulation of disability in MS can occur independently of relapse activity from early in the disease course. This scenario is underpinned by a more diffuse smouldering pathological process that may affect the entire CNS. Many putative pathological drivers of smouldering MS can be potentially modified by specific therapeutic strategies, an approach that may have major implications for the management of MS patients. We hypothesise that therapeutically targeting a state of 'no evident inflammatory disease activity' (NEIDA) cannot sufficiently prevent disability accumulation in MS, meaning that treatment should also focus on other brain and spinal cord pathological processes contributing to the slow loss of neurological function. This should also be complemented with a holistic approach to the management of other systemic disease processes that have been shown to worsen MS outcomes.
Despite therapeutic suppression of relapses, multiple sclerosis (MS) patients often experience subtle deterioration, which extends beyond the definition of "progression independent of relapsing activity." We propose the concept of smouldering-associated-worsening (SAW), encompassing physical and cognitive symptoms, resulting from smouldering pathological processes, which remain unmet therapeutic targets. We provide a consensus-based framework of possible pathological substrates and manifestations of smouldering MS, and we discuss clinical, radiological, and serum/cerebrospinal fluid biomarkers for potentially monitoring SAW. Finally, we share considerations for optimizing disease surveillance and implications for clinical trials to promote the integration of smouldering MS into routine practice and future research efforts.
Post-script: If you are an analyst and want more information on BTKi’s and MS, you cannot talk to me. I have too many conflicts of interest, so please don’t contact me.
Subscriptions and donations
MS-Selfie newsletters and access to the MS-Selfie microsite are free. In comparison, weekly off-topic Q&A sessions are restricted to paying subscribers. Subscriptions are being used to run and maintain the MS Selfie microsite, as I don’t have time to do it myself. You must be a paying subscriber if people want to ask questions unrelated to the Newsletters or Podcasts. If you can’t afford to become a paying subscriber, please email a request for a complimentary subscription (ms-selfie@giovannoni.net).
Important Links
X (Twitter) / LinkedIn / Medium
General Disclaimer
Please note that the opinions expressed here are those of Professor Giovannoni and do not necessarily reflect the positions of Queen Mary University of London or Barts Health NHS Trust. The advice is intended as general and should not be interpreted as personal clinical advice. If you have problems, please tell your healthcare professional, who will be able to help you.







Share this post